How I Found Out About My Genetic Variants And The Tools That I Used to Understand Them
This is a blog post about how I found out about the variants in my genome and the tools that I used to help me understand them.
I got whole genome testing done. I got my whole genome analyzed by Dante Labs and Sequencing which are companies that specialize in Next-generation Sequencing (NGS) which is a modern method of analyzing genetic material that allows for the rapid sequencing of large amount of DNA or RNA. My mother got her whole genome analyzed by Sequencing.
https://www.youtube.com/@DanteLabs
https://www.youtube.com/@sequencing
I obtained Binary Alignment Map (BAM) file and Variant Call Format (VCF) file from both Dante Labs and Sequencing.
https://en.wikipedia.org/wiki/Binary_Alignment_Map
https://en.wikipedia.org/wiki/Variant_Call_Format
https://www.youtube.com/watch?v=y4KqVfCdLo0
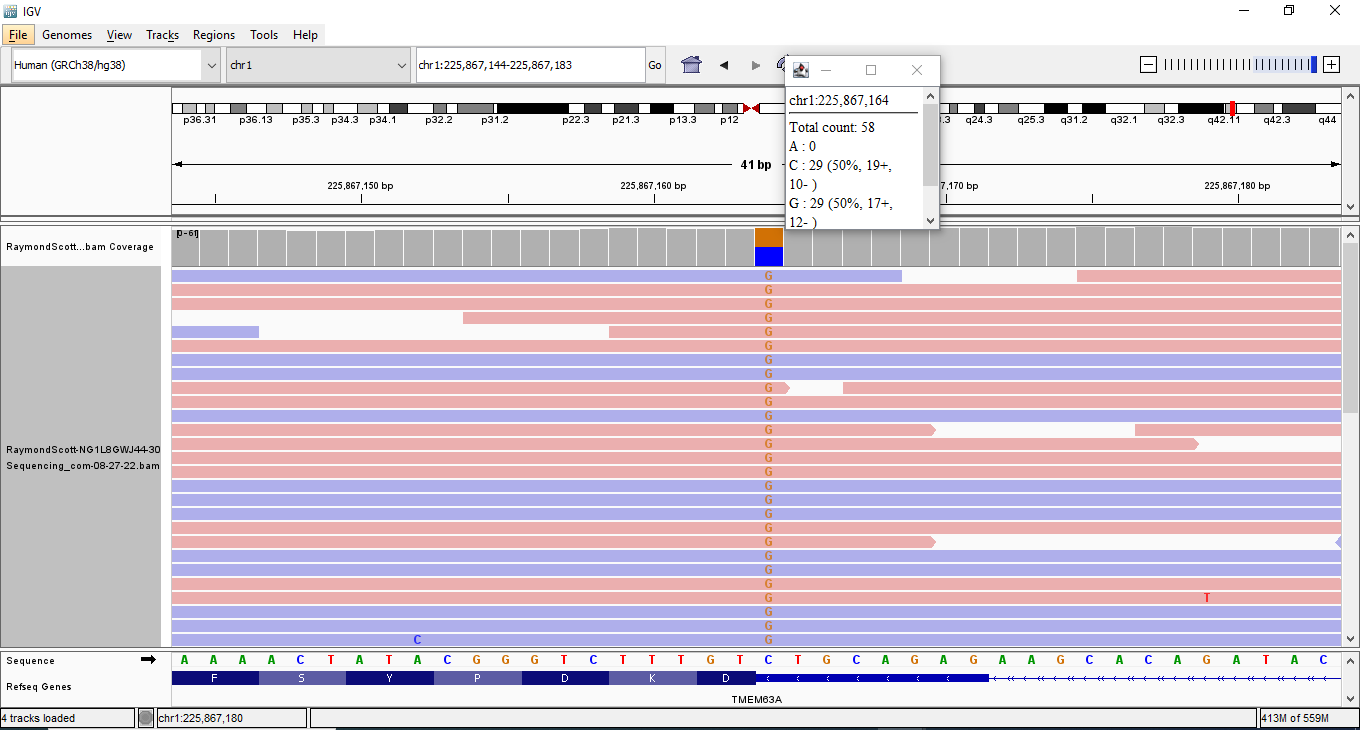
I used Integrative Genomics Viewer (IGV) for finding out variants and their chromosome location with the use of the BAM file and VCF file. If using VCF file, the rsID will also be found unless it's a novel variant. I check the read depth and quality score.
Integrative Genomics Viewer tutorial on youtube
https://www.youtube.com/playlist?list=PLSplvWwdPpSoyXjQ0xPs46CcA9Nzano9F
I found most of the variants by using my Dante Labs genomic data in Enlis Genome Personal which has a search filter. I can search by chromosome location, gene category or list, variation type, allele frequency, predicted deleterious (uses DANN In-Silico predictor) which is only available for Single Nucleotide Variants, tissue expression, protein impact, quality score, read depth, clinical significance, near gene start/end, zygosity, citations, trio analysis, mammalian conservation. It gives the gene location and rsID of the variant, but it also gives HGVS of a variant which is important to check out novel variants which don't have rsID numbers. The HGVS is useful for checking out variant in Ensembl's Variant Effect Predictor (VEP). Enlis Genome Personal uses HomoSapiensGRCh37 as the reference genome. Therefore, I use ClinGen allele registry and Genome Aggregation Database (gnomAD) browser to get the GRCh38 alignment information for the variants and overall verification. I used IGV to verify.
https://www.enlis.com/personal_edition.html
I click on the rsID number of the variant in Enlis Genome Personal, I get to the National Library of Medicine's Reference SNP report which has information about the organism, position, alleles, variation type, frequency, clinical significance, gene:consequence, publications, genomic view, HGVS, Submissions, History, and Flanks.
here is an example of one that I picked for my very rare MC4R Missense variant
https://www.ncbi.nlm.nih.gov/snp/rs52804924
I also found some variants from using Sequencing's Genome Explorer. I used IGV to verify.
https://sequencing.com/marketplace/genome-explorer-dna-data-search
I use the Genome Aggregation Database (gnomAD) browser to learn the transcripts, allele frequencies, and Combined Annotation Dependent Depletion (CADD) score of the variant. It will also show if it's reported in ClinVar.
https://gnomad.broadinstitute.org
Presentation - Use of the Genome Aggregation Database (gnomAD) (Anne O'Donnell-Luria)
https://www.youtube.com/watch?v=XdjjHdiVlrE&t=1204s
It also gives information about the GroupMax Filtering Allele Frequency FAF contains filtering allele frequency information from the genetic ancestry group with the highest FAF, not the filtering allele frequency information calculated on the genetic ancestry group with the highest AF. The filtering allele frequency (FAF) is the maximum credible genetic ancestry group AF (e.g. the lower bound of the 95% confidence interval (CI)). If the FAF is above the disease-specific threshold, then the observed AC is not compatible with pathogenicity.
https://gnomad.broadinstitute.org/help/faf
I've been referring to GroupMax Filtering Allele Frequency as Disease Allele Frequency. I caused some misunderstandings and offense by doing that. I rubbed some of my fellow neurodivergents the wrong way, and so I am now referring to it as Condition Allele Frequency. I have edited my blog posts and made changes.
I disregard variants that have condition allele frequencies in any population that are higher than the frequency of the condition for possible connections to a condition. In other words, I disregard the variant if the filtering allele frequency is above the condition-specific threshold.
I use a minimum CADD score of 20 for potential causal variants for rare conditions like Ataxia.
I use a minimum CADD score of 10 for potential risk factor variants for common conditions like Dyslexia, Dyspraxia, ADHD.
I use ClinGen Allele Registry
If I type in rsID, gnomAD, ClinVar RCV id, I can obtain the HGVS which I use to get information about a variant from Variant Effect Predictor (VEP)
https://reg.clinicalgenome.org/redmine/projects/registry/genboree_registry/landing
an example of a result
my very rare MC4R Missense variant
https://reg.clinicalgenome.org/redmine/projects/registry/genboree_registry/by_caid?caid=CA214149
I use VarSome to check out overall In-Silico Predictors to see if the variant is Benign or Pathogenic. I don't rely on just CADD score and DANN score. I disregard variants that show any predictions of Benign, Tolerated in any of the In-Silico predictors. The Varsome In-Silico predictors are available for only Single Nucleotide Variants.
I use Ensembl Variant Effect Predictor to check out the transcripts of the variant. This is useful for checking out novel variants. I use it to see if Stop Gained variants and Frameshift variants escape Nonsense Mediated Decay. Because Varsome's In-Silico predictors are unavailable for variants that aren't Single Nucleotide Variants, I checked Nonsense Mediated Decay escaping for the rare Frameshift variants. I use it to check for transcripts that involve regulatory features besides 5 Prime Untranslated Region (5'UTR) and 3 Prime Untranslated Region (3'UTR) like the Promoter. I use it to see if the 5'UTR transcript has upstream Open Reading Frames and Predicted consequences (uAUG Gained aka Premature Start Codon Gain, uAUG Lost aka Premature Start Codon Loss, uAUG Stop Gained aka Premature Stop Codon Gain, uStop Lost aka Premature Stop Codon Loss, uFrameshift) which In-Silico predictors don't take into account.
https://useast.ensembl.org/info/docs/tools/vep/index.html
I used Ensembl Variation - Calculated variant consequences page for information.
For each variant that is mapped to the reference genome, all overlapping Ensembl transcripts are identified. Ensembl use a rule-based approach to predict the effects that each allele of the variant may have on each transcript.The set of consequence terms, defined by the Sequence Ontology (SO), that can be currently assigned to each combination of an allele and a transcript is shown in the table below. It includes the SO term, SO description, SO acession, SO display term, and SO impact. It has diagram showing the location of each display term relative to the transcript structure:
https://grch37.ensembl.org/info/genome/variation/prediction/predicted_data.html
an example:
SO term: stop_gained
SO description: A sequence variant whereby at least one base of a codon is changed, resulting in a premature stop codon, leading to a shortened transcript
SO assession: SO:0001587 http://www.sequenceontology.org/miso/current_svn/term/SO:0001587
SO Display term: Stop gained
SO Impact: HIGH
Ensembl Training youtube channel that has training videos that I have been watching and learning from
https://www.youtube.com/@EnsemblHelpdesk
Introduction to Ensembl Genome Browser
https://www.youtube.com/playlist?list=PLqB8Yx1tGBMZrc1viF45x8ZuzkRmBPtGF
I use The Human Gene Database to learn about genes with information that includes aliases, disorders, domains, drugs, expression, function, genomics, localization, orthologs, paralogs, pathways, products, proteins, publications, sources, summaries, transcripts, variants, antibodies, assays, proteins, Inhib RNA, CRISPR, miRNA, Drugs, Animal Models, Cell Lines, Clones.
I also use malacards: The Human Disease Database. It is a searchable, integrated database that provides comprehensive, user-friendly information on all annotated human maladies. The knowledgebase automatically integrates disease-centric data from 75 selected web sources and is modeled on the architecture and richness of the popular GeneCards database of human genes.
I also use Mayaanlab to get information about the genes.
https://maayanlab.cloud/archs4/
https://maayanlab.cloud/Enrichr/#find


Comments