My Rare Frameshift Variants That Escape Nonsense-Mediated Decay
This is a blog post about my rare Frameshift variants. I focused only things only pertaining to the nervous system because of my interest in Developmental Neurogenomics as a neurodivergent with Dyslexia, Dyspraxia, ADHD. I also have Ataxia which is a rare neurological condition that involves coordination problems like Dyspraxia does.
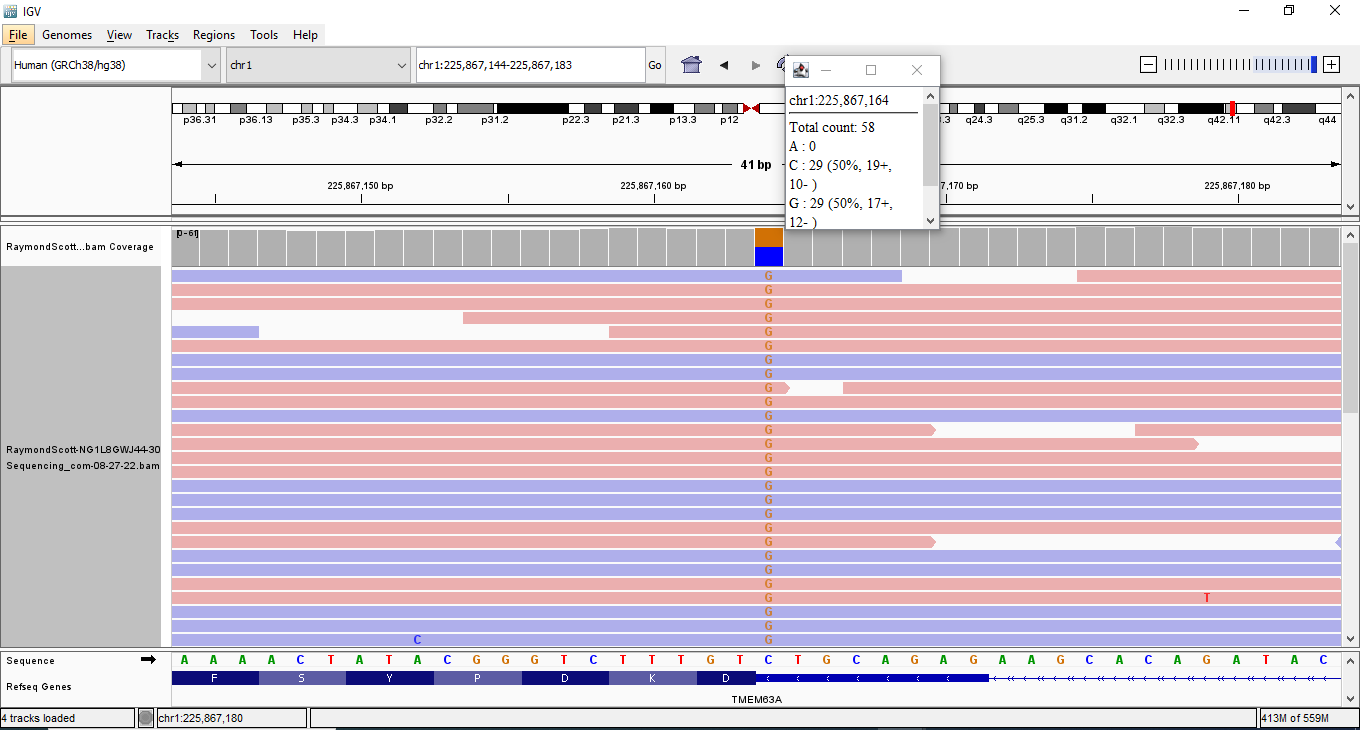
I used my Sequencing data
I used the Genome Aggregation Database (gnomAD) to check the allele frequencies, disease allele frequencies, and Combined Annotation Dependent Depletion (Scores). I consider only those with CADD scores of at least 20.
I used Ensembl's Variant Effect Predictor to see if they're predicted to escape Nonsense-Mediated Decay (NMD).
One-third of all inherited human diseases are caused by nonsense or frameshift mutations that introduce a premature stop codon in a transcript.
Nonsense-mediated RNA decay (NMD) is an evolutionarily conserved RNA quality control process that serves both as a mechanism to eliminate aberrant transcripts carrying premature stop codons, and to regulate expression of some normal transcripts.
Protein-truncating variants (PTVs) near the 3' end of genes may escape nonsense-mediated decay (NMD). PTVs in the NMD-escape region (PTVescs) can cause Mendelian disease but are difficult to interpret given their varying impact on protein function.
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC10534055/
https://www.nature.com/articles/s41467-020-17526-5
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC10685860/
https://pubmed.ncbi.nlm.nih.gov/38091987/
Checking out only rare frameshift variants that are Nonsense-Mediated Decay escaping. I have three. Two have disease allele frequencies under 0.01% which is the maximum disease allele frequency that I consider for Ataxia. Those are DOK7 variant and GPNMB variant.
Two of the the variants involve regulatory features. DOK7 variant and CEP290 variant involve 3 Prime Untranslated Region.
Two of the variants are reported in ClinVar. DOK7 variant is reported as Pathogenic for Congenital Myasthenic Syndrome and Fetal Akinesia Deformation Sequence, but they are Autosomal Recessive conditions. CEP290 variant is reported as Uncertain Significance for Familial aplasia of the vermis, Meckel-Gruber syndrome, Nephronophthisis, Leber congenital amaurosis, CEP290-Related Disorder.
GPNMB has a variant associated with Ataxia Telangiectasia phenotype and a variant associated with Hyperactive behavior phenotype. Knockdown of GPNMB results in hyperactivity phenotype.
CEP290 has variants associated with Cerebellar Ataxia and Autism
https://neurodivergence.blogspot.com/2024/03/my-rare-single-nucleotide-variants-that.html
rs606231133 4-3493358-GC-G no data available for my mother
DOK7
6 out of 1,596,906 (0.0003757%) Condition allele frequency is 0.001752%
24.0
Reported in ClinVar as Pathogenic for Congenital Myasthenic Syndrome and Fetal Akinesia Deformation Sequence

DOK7 (Docking Protein 7) is a Protein Coding gene. Diseases associated with DOK7 include Myasthenic Syndrome, Congenital, 10 and Fetal Akinesia Deformation Sequence 3. Gene Ontology (GO) annotations related to this gene include protein kinase binding and insulin receptor binding.
The protein encoded by this gene is essential for neuromuscular synaptogenesis. The protein functions in aneural activation of muscle-specific receptor kinase, which is required for postsynaptic differentiation, and in the subsequent clustering of the acetylcholine receptor in myotubes. This protein can also induce autophosphorylation of muscle-specific receptor kinase. Mutations in this gene are a cause of familial limb-girdle myasthenia autosomal recessive, which is also known as congenital myasthenic syndrome type 1B. Alternative splicing results in multiple transcript variants. [provided by RefSeq, Sep 2009]
https://www.genecards.org/cgi-bin/carddisp.pl?gene=DOK7
Enrichr
Jensen TISSUES: Brain (Cerebellum, Cerebral Cortex, Hippocampus), motorneuron
Knockdown of dok7 results in abnormal gait MP:0001406 phenotype.
Knockdown of dok7 results in abnormal neuromuscular synapse morphology MP:0001053 phenotype.
dok7 is found in Cerebellar hypoplasia (HP:0001321).
dok7 is found in Abnormality of the neuromuscular junction (HP:0003398).
dok7 is found in Waddling gait (HP:0002515).
dok7 is found in Hydrocephalus (HP:0000238)
https://maayanlab.cloud/Enrichr/#find!gene=DOK7
GPNMB
40 out of 1,614,080 (0.002478%) Condition allele frequency is 0.002313%
23.7

GPNMB (Glycoprotein Nmb) is a Protein Coding gene. Diseases associated with GPNMB include Amyloidosis, Primary Localized Cutaneous, 3 and Lichen Amyloidosis. Among its related pathways are Signaling by PTK6 and Adhesion. Gene Ontology (GO) annotations related to this gene include heparin binding and integrin binding. An important paralog of this gene is PMEL.
The protein encoded by this gene is a type I transmembrane glycoprotein which shows homology to the pMEL17 precursor, a melanocyte-specific protein. GPNMB shows expression in the lowly metastatic human melanoma cell lines and xenografts but does not show expression in the highly metastatic cell lines. GPNMB may be involved in growth delay and reduction of metastatic potential. Two transcript variants encoding different isoforms have been found for this gene. [provided by RefSeq, Jul 2008]
https://www.genecards.org/cgi-bin/carddisp.pl?gene=GPNMB
Enrichr
Jensen TISSUES: is expressed in Brain (Cerebellum, Cerebral cortex, Corpus callosum, Frontal lobe, Hypothalamus, Parietal lobe, Pineal gland, Temporal lobe), spinal cord
GPNMB mapped to the strongest SNP related to Parkinson's Disease.
GPNMB mapped to the strongest SNP related to Parkinson's Disease Or First Degree Relation To Individual With Parkinson's Disease.
GPNMB mapped to the strongest SNP related to Cerebrospinal Fluid Biomarker Levels.
Variant in GPNMB is associated with Alzheimer Disease, Late Onset phenotype.
Variant in GPNMB is associated with Alzheimer's Disease phenotype.
Variant in GPNMB is associated with r phenotype.
Variant in GPNMB is associated with Brain Ischemia phenotype.
Variant in GPNMB is associated with Cerebral Infarction phenotype.
Variant in GPNMB is associated with Cerebral Ischemia phenotype.
Variant in GPNMB is associated with Hyperactive behavior phenotype.
Variant in GPNMB is associated with Motor neuron atrophy phenotype.
Variant in GPNMB is associated with Neurodegenerative Disorders phenotype.
Variant in GPNMB is associated with Parkinson Disease phenotype.
Variant in GPNMB is associated with Pituitary Adenoma phenotype.
Knockdown of GPNMB results in MP0003633 abnormal nervous system phenotype.
Knockdown of GPNMB results in MP:0011396 abnormal sleep behavior phenotype.
Knockdown of GPNMB results in MP0002229 neurodegeneration phenotype.
Knockdown of GPNMB results in MP0002752 abnormal somatic nervous phenotype.
Knockdown of GPNMB results in MP0002882 abnormal neuron morphology phenotype.
Knockdown of GPNMB results in MP:0001399 hyperactivity phenotype.
Knockdown of GPNMB results in MP:0011396 abnormal sleep behavior phenotype.
https://maayanlab.cloud/Enrichr/#find!gene=GPNMB

Enrichr
Jensen TISSUES: is expressed in Brain (Cerebellum, Cerebral cortex, Corpus callosum, Frontal lobe, Hypothalamus, Parietal lobe, Pineal gland, Temporal lobe), spinal cord
Variant in GPNMB is associated with Autistic Spectrum Disorders phenotype
Variant in CEP290 is associated with Autistic behavior phenotype
Variant in CEP290 is associated with Autistic disorder phenotype
Variant in CEP290 is associated with Cerebellar Ataxia phenotype
Variant in CEP290 is associated with Cerebellar cyst phenotype
Variant in CEP290 is associated with Cerebellar vermis aplasia or hypoplasia phenotype
Variant in CEP290 is associated with Cerebellar vermia hypoplasia phenotype
Variant in CEP290 is associated with Epilepsy phenotype
Knockdown of CEP290 results in MP:0000849 abnormal cerebellum morphology phenotype
Knockdown of CEP290 results in MP:000064 abnormal cerebellum vermis morphology phenotype
Knockdown of CEP290 results in MP000632 abnormal nervous system phenotype
Knockdown of CEP290 results in MP0002abnormal somatic nervous phenotype.
Knockdown of CEP290 results in MP0002882 abnormal neuron morphology phenotype.
https://maayanlab.cloud/Enrichr/#find!gene=CEP290
.png)


Comments