My Rare Single Nucleotide Variants that are shown to be potentially Pathogenic according to In-Silico Predictors.
This is a blog post about my rare Single Nucleotide Variants that are shown to be potentially Pathogenic according to In-Silico Predictors. I focused only things only pertaining to the nervous system because of my interest in Developmental Neurogenomics as a neurodivergent with Dyslexia, Dyspraxia, ADHD. I also have Ataxia which is a rare neurological condition that involves coordination problems like Dyspraxia does.
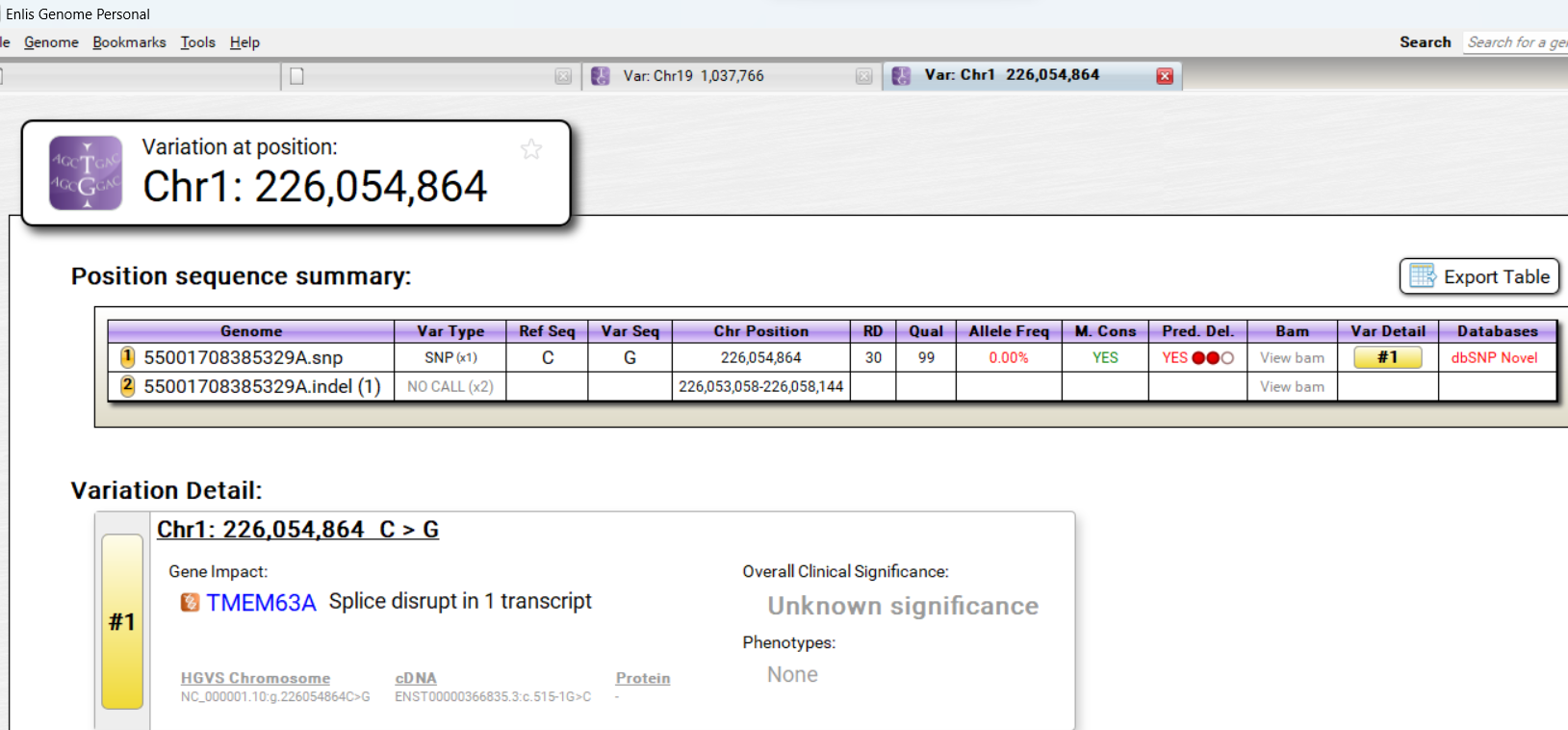
I used my Dante Lab data in Enlis Genome Personal.
I searched for variants with frequences of less than 0.1% and predicted Deleterious according to DANN.
Then I used the Genome Aggregation Database (gnomAD) to check the allele frequencies, disease allele frequencies, and Combined Annotation Dependent Depletion (Scores). I consider only those with CADD scores of at least 20.
Then I checked to see In Silico Predictions at Varsome.com. I considered only variants that have Meta cores that show potential to be Pathogenic and show no sign of being benign.
Meta scores: These predictors determine a pathogenicity based on the combined evidence from multiple other in-silico predictors. Note: Engines are assigned a prediction points score based on the strength of the calibrated prediction. Supporting: 1 point. Moderate: 2 points. Strong: 4 points. Very Strong: 8 points.
BayesDel addAF
A deleteriousness prediction meta-score for SNVs and indels with inclusion of MaxAF. See https://doi.org/10.1002/humu.23158 for details. The range of the score in dbNSFP is from -1.11707 to 0.750927. The higher the score, the more likely the variant is pathogenic. The author suggested cutoff between deleterious ('D') and tolerated ('T') is 0.0692655.
BayesDel noAF
A deleteriousness prediction meta-score for SNVs and indels without inclusion of MaxAF. See https://doi.org/10.1002/humu.23158 for details. The range of the score in dbNSFP is from -1.31914 to 0.840878. The higher the score, the more likely the variant is pathogenic. The author suggested cutoff between deleterious ('D') and tolerated ('T') is -0.0570105.
REVEL
The Rare Exome Variant Ensemble Learner REVEL is an ensemble method for predicting the pathogenicity of missense variants. It integrates scores from MutPred, FATHMM v2.3, VEST 3.0, PolyPhen-2, SIFT, PROVEAN, MutationAssessor, MutationTaster, LRT, GERP++, SiPhy, phyloP, and phastCons. Score range from 0 to 1 and variants with higher scores are predicted to be more likely to be pathogenic.
REVEL does not provide a descriptive prediction but for convenience, we display scores above 0.5, as 'likely disease causing' and display scores below 0.5 as 'likely benign'. It was estimated that 75.4% of disease mutations but only 10.9% of neutral variants have a score above 0.5
We strongly recommend the actual score is used when assessing a variant and a cut-off appropriate to your requirements is chosen.
MetaLR
MetaLR is an ensemble score using Logistic Regression (LR) to integrate nine prediction scores (SIFT, PolyPhen-2, GERP++, MutationTaster, Mutation Assessor, FATHMM, LRT, SiPhy and PhyloP) and allele frequencies in 1KG database. The tool has been trained on 36,192 SNVs from UniProt. Range 0 to 1.
MetaRNN
Our recurrent neural network (RNN) based ensemble prediction score, which incorporated 16 scores (SIFT, Polyphen2_HDIV, Polyphen2_HVAR, MutationAssessor, PROVEAN, VEST4, M-CAP, REVEL, MutPred, MVP, PrimateAI, DEOGEN2, CADD, fathmm-XF, Eigen and GenoCanyon), 8 conservation scores (GERP, phyloP100way_vertebrate, phyloP30way_mammalian, phyloP17way_primate, phastCons100way_vertebrate, phastCons30way_mammalian, phastCons17way_primate and SiPhy), and allele frequency information from the 1000 Genomes Project (1000GP), ExAC, and gnomAD. Larger value means the SNV is more likely to be damaging. Scores range from 0 to 1.
MetaSVM
MetaSVM is an ensemble score using Support Vector Machine (SVM) to integrate nine prediction scores (SIFT, PolyPhen-2, GERP++, MutationTaster, Mutation Assessor, FATHMM, LRT, SiPhy and PhyloP) and allele frequencies in 1KG database. The tool has been trained on 36,192 SNVs from UniProt. Range -2.0058 to +3.0399.
All variants have allele frequencies that are under 0.01%. All of them have disease allele frequencies that are under 0.01% which is the maximum disease allele frequency that I consider for Ataxia.
Two of the variants are a Stop Gained in SGSM2 and Splice Acceptor in TMEM63A which are High Impact variants. The other three are Missense variants in MC4R, IFT172, and SLC4A2 which are Moderate Impact variants.
Two of the five variants involve regulatory features. TMEM63A variant involves Enhancer. IFT172 variant involves 3 Prime Untranslated Region.
Two of the five variants are reported in ClinVar. MC4R variant is reported as Pathogenic for Obesity. SLC4A2 is reported as Uncertain Significance for Inborn Genetic Disease.
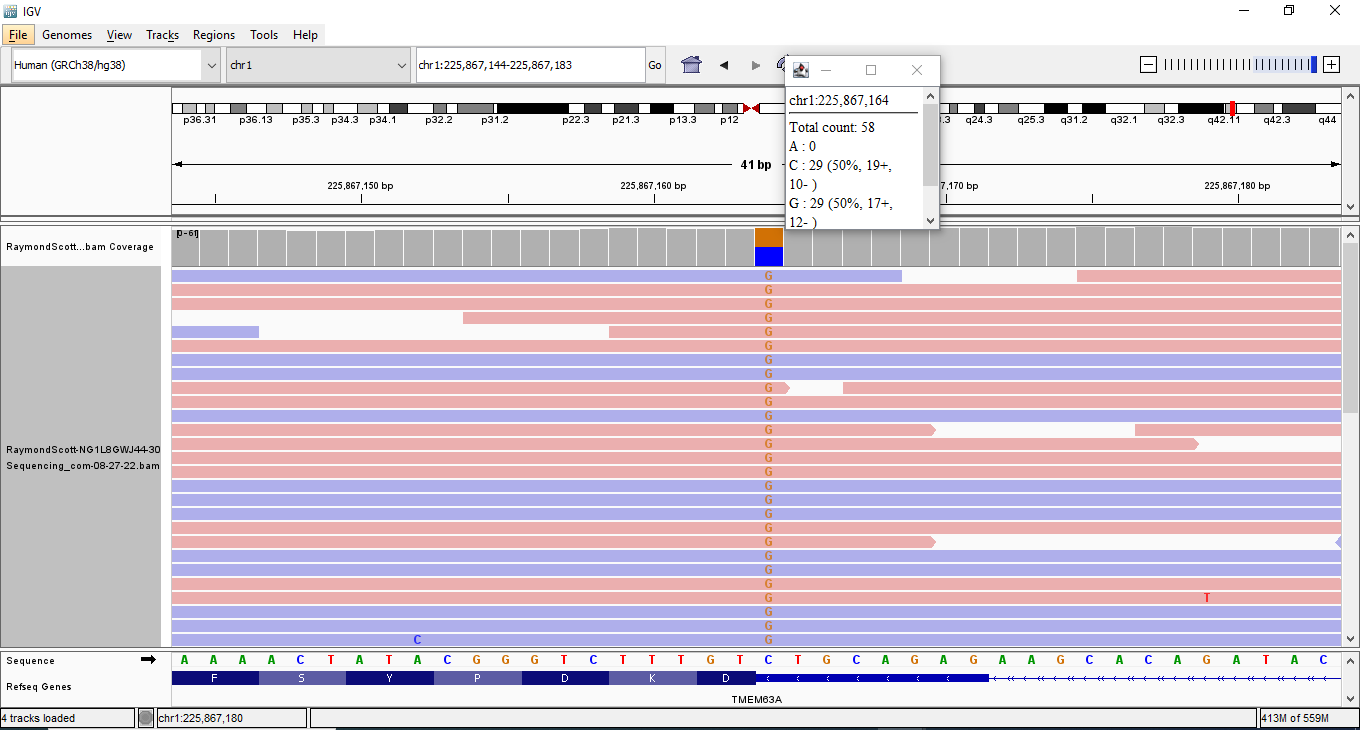
My Sequencing Genomic data variants in Integrative Genomics Viewer (IGV)

rs867998099 17-2379146-C-T no data available for my mother
Stop Gained
SGSM2
3 out of 1,614,054 (0.0001859%) Condition allele frequency is 0.00028%
40.0
https://reg.clinicalgenome.org/redmine/projects/registry/genboree_registry/by_caid?caid=CA286906028
https://maayanlab.cloud/archs4/gene/SGSM2
Jensen TISSUES: is expressed in Brain (Amygala Caudate nucleus, Cerebellum, Cerebral cortex, Cerebral peduncle, Cingulate cortex, Embryonic brain, Frontal lobe, Hippocampus, Hypothalamus, Medulla Oblongata, Occipital lobe, Parietal lobe, Pons, Prefontal cortex, Subthalamic nucleus, Superior cervical ganglion, Temporal lobe, Thalamus, Thymus), Spinal cord, Spinal ganglion, Superior cervical ganglion, Trigeminal ganglion
SGSM2 is predicted to be associated with Cerebellar ataxia and hypogonadotropic hypogonadism.
SGSM2 is predicted to be associated with Cluttering.
SGSM2 is predicted to be associated with Fatty acid hydroxylase-associated neurodegeneration.
SGSM2 is predicted to be associated with Hypothalamic dysfunction.


rs966645435 1-225867164-C-G my mother has it too
Splice Acceptor, Enhancer
TMEM63A
18 out of 1,614,038 (0.001115%) Condition allele frequency is 0.001063%
32.0
https://reg.clinicalgenome.org/redmine/projects/registry/genboree_registry/by_caid?caid=CA38540925
3 Pathogenic Moderate
TMEM63A (Transmembrane Protein 63A) is a Protein Coding gene. Diseases associated with TMEM63A include Leukodystrophy, Hypomyelinating, 19, Transient Infantile and Hypomyelinating Leukodystrophy. Among its related pathways are Innate Immune System. Gene Ontology (GO) annotations related to this gene include nucleotide binding. An important paralog of this gene is TMEM63B.
Enables mechanosensitive ion channel activity. Predicted to be involved in cation transmembrane transport. Located in centriolar satellite and lysosomal membrane. Implicated in hypomyelinating leukodystrophy. [provided by Alliance of Genome Resources, Apr 2022]
https://www.genecards.org/cgi-bin/carddisp.pl?gene=TMEM63A
Human Phenotypes: Ataxia, Dysmetria, Specific learning disability, Intention tremor, Delayed gross motor development, Abnormality of movement, Tremor, Abnormality of coordination, Tremor, Motor delay, Abnormality of the nervous system, Abnormal central motor function, Action tremor, Abnormality of eye movement, Neurodevelopmental abnormality, Neurodevelopmental delay, Abnormal nervous system physiology, Kinetic tremor, Nystagamus
https://www.genecards.org/cgi-bin/carddisp.pl?gene=TMEM63A
Intention tremor, Abnormal auditory evoked potentials, Slow saccadic eye movements, Spastic gait, Dysmetria, Clumsiness
https://maayanlab.cloud/archs4/gene/TMEM63A
Enrichr
Jensen TISSUES: is expressed in Brain (Cerebral Cortex, Frontal lobe, Hypothalamus, Medulla oblongata, Occipital lobe, Parietal lobe, Pons, Subthalamic nucleus, Thalamus), Spinal cord
TMEM63A is predicted to be associated with Fatty acid hydroxylase-associated neurodegeneration.
https://maayanlab.cloud/Enrichr/#find!gene=TMEM63A

rs52804924 18-60371454-G-T my mother has it too
Missense
MC4R
34 out of 1,605,580 (0.002107%) Condition allele frequency is 0.004374%
27.9
Reported in ClinVar as Pathogenic, Likely Pathogenic for Obesity
https://reg.clinicalgenome.org/redmine/projects/registry/genboree_registry/by_caid?caid=CA214149
20 Pathogenic Very Strong
MC4R (Melanocortin 4 Receptor) is a Protein Coding gene. Diseases associated with MC4R include Body Mass Index Quantitative Trait Locus 20 and Obesity Due To Melanocortin 4 Receptor Deficiency. Among its related pathways are Class A/1 (Rhodopsin-like receptors) and GPCR downstream signalling. Gene Ontology (GO) annotations related to this gene include G protein-coupled receptor activity and peptide hormone binding. An important paralog of this gene is MC5R.
The protein encoded by this gene is a membrane-bound receptor and member of the melanocortin receptor family. The encoded protein interacts with adrenocorticotropic and MSH hormones and is mediated by G proteins. This is an intronless gene. Defects in this gene are a cause of autosomal dominant obesity. [provided by RefSeq, Jan 2010]
https://www.genecards.org/cgi-bin/carddisp.pl?gene=MC4R
Human Phenotypes: Visual hallucinations, Delayed gross motor development, Agitation, Progressive cerebellar ataxia, Anxiety, Broad-based gait, Truncal ataxia, Dysdiadochokinesis, Psychosis, Dysmetria, Impaired smooth pursuit, Gait imbalance, Depression, Restlessness, Action tremor, Delusions, Specific learning disability, Impaired social interactions, Abnormal social behavior, Abnormality of pain sensation, Abnormality of ocular pursuit, Postural instability, Poor eye contact
https://maayanlab.cloud/archs4/gene/MC4R
Jensen TISSUES: is expressed in Brain (Cerebellum, Cerebral Cortex, Hypothalamus), Autonomic nervous system, Ganglion
Knockdown of MC4R results in MP0004924 abnormal behavior phenotype.
Knockdown of MC4R results in MP0002066 abnormal motor capabilities phenotype.


rs976552782 2-27480144-C-A no data available for my mother
Missense, 3'UTR
IFT172
7 out of 1,613,130 (0.0004339%) Condition allele frequency is 0.003474%
26.6
https://reg.clinicalgenome.org/redmine/projects/registry/genboree_registry/by_caid?caid=CA44515401
14 Pathogenic Very Strong
IFT172 (Intraflagellar Transport 172) is a Protein Coding gene. Diseases associated with IFT172 include Short-Rib Thoracic Dysplasia 10 With Or Without Polydactyly and Retinitis Pigmentosa 71. Among its related pathways are Signaling by Hedgehog and Organelle biogenesis and maintenance. Gene Ontology (GO) annotations related to this gene include binding. An important paralog of this gene is IFT140.
This gene encodes a subunit of the intraflagellar transport subcomplex IFT-B. Subcomplexes IFT-A and IFT-B are necessary for ciliary assembly and maintenance. Mutations in this gene have been associated with skeletal ciliopathies, with or without polydactyly, such as such short-rib thoracic dysplasias 1, 9 or 10. [provided by RefSeq, Mar 2014]
https://www.genecards.org/cgi-bin/carddisp.pl?gene=IFT172
Human Phenotypes: Gait imbalance, Dysmetric saccades, Postural instability, Gaze-evoked nystagmus, Abnormality of ocular smooth pursuit, Action tremor, Hyperacusis, Progressive cerebellar ataxia, Insomnia, Poor coordination, Visual hallucinations, Impaired smooth pursuit, Dysdiadochokinesis, Abnormality of saccadic eye movements, Depression, Oculomotor apraxia
https://maayanlab.cloud/archs4/gene/IFT172
Enrichr
Jensen TISSUES: is expressed in Brain (Cerebellum, Cerebral Cortex, Corpus callosum, Frontal lobe, Hippocampus, Hypothalamus, Occipital lobe, Parietal lobe, Temporal lobe) , Spinal cord
Knockdown of IFT172 results in MP:0000913 abnormal brain development phenotype.
Knockdown of IFT172 results in MP0003632 abnormal nervous system phenotype.
Knockdown of IFT172 results in MP:0000929 open neural tube phenotype.
Knockdown of IFT172 results in MP:0002151 abnormal neural tube morphology phenotype.
Knockdown of IFT172 results in MP0000955 abnormal spinal cord phenotype.
Knockdown of IFT172 results in MP0002882 abnormal neuron morphology phenotype.
Knockdown of IFT172 results in MP:0004261 abnormal embryonic neuroepithelium morphology phenotype.
Knockdown of IFT172 results in MP:0011380 enlarged brain ventricles phenotype.
Knockdown of IFT172 results in MP0002152 abnormal brain morphology phenotype.


rs746816423 7-151072063-G-A my mother has it too
Missense
SLC4A2
23 out of 1,613,906 (0.0014125%) Condition allele frequency is same
28.2
Jensen TISSUES: is expressed in Brain (Cerebellum, Cerebral cortex, Corpus callosum, Frontal lobe, Hippocampus, Hypothalamus, Occipital lobe, Parietal lobe, Temporal lobe), Spinal cord
Knockdown of SLC4A2 results in MP:0001393 ataxia phenotype.


.png)


{kind=link}
{kind=link}
Comments