My Neurological Makeup includes both Neurodivergence and Ataxia.
I am not just a neurodivergent with Dyslexia, Dyspraxia, ADHD. My neurological make include both Neurodivergence and Ataxia. Ataxia is defined as the inability to generate coordinated voluntary movement, which can manifest clinically with signs and symptoms such as clumsiness, nystagmus, dysmetria, abnormal or unsteady gait, dysdiadochokinesis, or dyssynergia. It's a rare neurological problem. The prevalence rate for hereditary ataxias is 10 cases per 100,000 individuals. The prevalence rate for childhood ataxias is 26 cases per 100,000 children.
I noticed Ataxia listed on my Veteran Affairs (VA) Problem List. I thought my coordination problems were connected to Dyspraxia which is a common neurodivergent condition that commonly occurs with Dyslexia and Attention Deficit-Hyperactivity Disorder which are other neurodivergent conditions that I have. I had noticeable coordination problems in early childhood which led to neurological testing being done on me, and so I have history of ataxia in childhood. My mother told me that my father had problems with coordination like I have, and so it seems that I inherited from him. He also had problems with speech, auditory processing, and learning like I have. Unlike me, he had problems with perceiving and understanding emotional cues which make me suspect that he was on the Autistic Spectrum. He was also a highly sensitive person like my mother and me. My paternal Aunt Carrie told me that my father was hyperactive. I inherited my neurodivergence from both my parents.
I have other coordination issues that are listed on my VA Problem List. They are Saccadic Eye Movement deficiencies and Smooth Pursuit movement deficiencies, and those are all eye coordination issues. Dr Harold Levinson's testing included testing my saccadic and smooth pursuit movements, and I was found to be abnormal on both of them. He also tested my optokinetic kinetic eye movement, and I was found to be abnormal on it. The VA neurologists and Dr. Levinson recorded that I had dysdiadochokinesis. The VA psychoneurologist noted that my speech is mildly Dysarthric. I have a soft, high pitched voice.
The VA neurologists recorded that I had abnormal cerebellar system that included
Nystagamus optokinetic with slowed saccades but symmetric horizontal vertically difficulty initiating saccades to command and read.
Dysdiadochokinesia present symmetrically all 4 extremities.
Ataxia on target finger-nose but dysmetria with moving target showing slowed saccadic movements.
Gait with mild ataxia
The VA neurologists confirmed my Dyslexia and Dyspraxia, and they noted that it was genetic and not acquired for I had no progression of symptoms. My VA Problems list include Dyslexia and Abnormal Auditory Perception. I had early therapies in childhood that included auditory therapy, speech therapy, phonics training, and motor skills therapy. I was in special education in my early elementary school years, and I was even mistakenly identified as having an intellectual disability which led to mother getting her Fallopian Tubes tied because she thought that I would need a lot of attention and care. Dr. Levinson diagnosed me as having Cerebellar-Vestibular Dysfunction which he claims is the root of Dyslexia and other neurodivergent conditions which he refers as being all part of Dyslexic Syndrome. The truth is that my coordination problems aren't from just dyspraxia alone. They're also from Ataxia. I can relate to the Dyspraxia because of the difficulty with coordination, planning, learning movements, problems processing a sequence of actions/following multi-step instructions, poor working/short term memory, poor organization, but there is strong overlap and co-morbidity with Dyslexia and ADHD. There is also strong overlap with autism. A lot of people with Dyspraxia have Aspergers Syndrome. I don't have social interaction impairments,and so I am not autistic. The Veteran Affairs neurologists noted that during their examination of me.
I felt that I had to set the record straight that my neurological processing issues aren't just from neurodivergent conditions but from serious cerebellar problems that had been addressed in early childhood but never went away. They became mild and subtle as the result of early childhood therapies. The problems were always present but misunderstood which led to wrong judgments about me by others and myself as well as psychiatric misdiagnoses. I want to help raise awareness and understanding of people with ataxia and that there are neurodivergents that have ataxia.
My neuropsychological testing and neurological testing 2004-2006
Psychologist Dr. McMahon in 2004, Dr. Levinson in 2005, Veteran Affairs Neurologists and Neuropsychologist in 2006
https://neurodivergence.blogspot.com/2021/01/my-neurodivergence-testing.html
Information about Ataxia
https://www.americanbrainfoundation.org/diseases/ataxia/
https://www.sciencedirect.com/science/article/abs/pii/S1751722219301738
https://www.ataxia.org/what-is-ataxia/
https://www.childrens.com/specialties-services/conditions/ataxia
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5347818/
My VA Problem List


My possible genetic indicators for Ataxia
I have done whole genome testing through Dante Labs and Sequencing. I have developed a very strong interest in Genomics/Genetics including especially Neurogenetics and genomic studies of neurodivergent conditions.
Considering only my rare variants with allele frequences of no more 0.02%, genes in connection to my Ataxia: SGSM2, IFT172, MC4R, SLC4A2, TMEM63A, TRPM8, CEP290, CYLD, PES1.
I inherited the MC4R, TMEM63A, SLC4A2, CEP290, and PES1 variants from my mother.
There is insufficient data for my mother to see if any other variants are inherited from her.
I never knew my father who has been dead since 1993, and so I have no way of knowing what variants that I inherited from him.
I looked at variants in genes that have ataxia as human phenotype and/or expression in cerebellar system
I included Human phenotypes that have to do with both neurological disorders and psychiatric disorders. There is a strong overlap between them that needs to be considered. My own neurological problems were undetected by mental health professionals throughout my life with me having very little understanding of myself. The Navy doctors and psychologist didn't know what was really going on with me.

What is Combined Annotation Dependent Depletion (CADD)?
CADD is a tool for scoring the deleteriousness of single nucleotide variants as well as insertion/deletions variants in the human genome.
While many variant annotation and scoring tools are around, most annotations tend to exploit a single information type (e.g. conservation) and/or are restricted in scope (e.g. to missense changes). Thus, a broadly applicable metric that objectively weights and integrates diverse information is needed. Combined Annotation Dependent Depletion (CADD) is a framework that integrates multiple annotations into one metric by contrasting variants that survived natural selection with simulated mutations.
C-scores strongly correlate with allelic diversity, pathogenicity of both coding and non-coding variants, and experimentally measured regulatory effects, and also highly rank causal variants within individual genome sequences. Finally, C-scores of complex trait-associated variants from genome-wide association studies (GWAS) are significantly higher than matched controls and correlate with study sample size, likely reflecting the increased accuracy of larger GWAS.
CADD can quantitatively prioritize functional, deleterious, and disease causal variants across a wide range of functional categories, effect sizes and genetic architectures and can be used prioritize causal variation in both research and clinical settings.
In addition to this website, CADD has been described in three publications. The most recent manuscript describes CADD-Splice (CADD v1.6), the latest extension of CADD to improve its predictions of splicing effects:
https://cadd.gs.washington.edu/
What are the scores in the files and which score cutoff should I use?
The last column of the provided files is the PHRED-like (-10*log10(rank/total)) scaled C-score ranking a variant relative to all possible substitutions of the human genome (8.6x10^9). Like explained above, a scaled C-score of greater of equal 10 indicates that these are predicted to be the 10% most deleterious substitutions that you can do to the human genome, a score of greater or equal 20 indicates the 1% most deleterious and so on.
The second to last column is the raw score of the model. Due to the high mislabeling in our training data, it does not have any interpretation (even the sign does not have an interpretation). The higher the raw C score the more predicted to be deleterious. If you want to do a non-parametric test between sets of variants, we recommend using this raw C-score (see above). If you want to put a cutoff on deleteriousness, we recommend the last column (scaled C-score) as it has some interpretation by relating the raw C-score to the raw C-scores of all possible substitutions in the human genome.
If you would like to apply a cutoff on deleteriousness, e.g. to identify potentially pathogenic variants, we would suggest to put a cutoff somewhere between 10 and 20. Maybe at 15, as this also happens to be the median value for all possible canonical splice site changes and non-synonymous variants in CADD v1.0. However, there is not a natural choice here -- it is always arbitrary. We therefore recommend integrating C-scores with other evidence and to rank your candidates for follow up rather than hard filtering.
https://cadd.gs.washington.edu/info
I checked for Single Nucleotide Variants with an allele frequency of no more than 0.02%, predicted to be deleterious according to DANN in Enlis Genome Personal, and have CADD scores of at least 20. My Dante Labs genomic data is in Enlis Genome Personal. I checked to see if the variants also appeared in my Sequencing genomic data in Genome Explorer. Dante Labs genome can have variants that Sequencing don't have and vice versa.
I checked them in Varsome, and I disqualified any that have any predictions of being Benign/Likely Benign, Tolerated.
One is a Stop Gained. Two are Missense. One is a Splice Acceptor. Stop Gained and Splice Acceptor are High Impact variants that can lead to loss of function.
Stop Gained - A sequence variant whereby at least one base of a codon is changed, resulting in a premature stop codon, leading to a shortened polypeptide.

Summary: Annotating genetic variants, especially non-coding variants, for the purpose of identifying pathogenic variants remains a challenge. Combined annotation-dependent depletion (CADD) is an algorithm designed to annotate both coding and non-coding variants, and has been shown to outperform other annotation algorithms. CADD trains a linear kernel support vector machine (SVM) to differentiate evolutionarily derived, likely benign, alleles from simulated, likely deleterious, variants. However, SVMs cannot capture non-linear relationships among the features, which can limit performance. To address this issue, we have developed DANN. DANN uses the same feature set and training data as CADD to train a deep neural network (DNN). DNNs can capture non-linear relationships among features and are better suited than SVMs for problems with a large number of samples and features. We exploit Compute Unified Device Architecture-compatible graphics processing units and deep learning techniques such as dropout and momentum training to accelerate the DNN training. DANN achieves about a 19% relative reduction in the error rate and about a 14% relative increase in the area under the curve (AUC) metric over CADD’s SVM methodology.
https://academic.oup.com/bioinformatics/article/31/5/761/2748191rs867998099 C/T no data available for my mother
Stop Gained
SGSM2
3 out of 1,614,054 (0.0001859%)
DANN: 0.9978386290023271
CADD: 40.0
Metascore 8 Pathogenic very strong
https://reg.clinicalgenome.org/redmine/projects/registry/genboree_registry/by_caid?caid=CA286906028
https://gnomad.broadinstitute.org/variant/17-2379146-C-T?dataset=gnomad_r4
https://varsome.com/variant/hg38/rs867998099?annotation-mode=germline
Human Phenotypes: Progressive cerebellar ataxia, Depression, Depression, Poor eye contact, Postural instability, Impaired social interactions, Abnormal social behavior, Dysdiadochokinesis, Broad-based gait, Insomnia, Gaze-evoked nystagmus, Anxiety, Impaired smooth pursuit, Dysmetric saccades, Dysmetria, Truncal ataxia, Spastic gait, Agitation, Abnormality of ocular smooth pursuit, Intention tremor, Hyperacusis
https://maayanlab.cloud/archs4/gene/SGSM2
.png)


Missense
MC4R
34 out of 1,605,580 (0.002107%)
https://gnomad.broadinstitute.org/variant/18-60371454-G-T?dataset=gnomad_r4
Reported Pathogenic, Likely Pathogenic for Obesity in Clinvar



rs976552782 C/A no data available for my mother
Missense
IFT172
7 out of 1,613,130 (0.0004339%)
DANN: 0.9981074046322775
CADD: 26.6
PP3: Pathogenic Moderate
Metascore 14 Pathogenic very strong
https://reg.clinicalgenome.org/redmine/projects/registry/genboree_registry/by_caid?caid=CA44515401
https://gnomad.broadinstitute.org/variant/2-27480144-C-A?dataset=gnomad_r4
https://varsome.com/variant/hg38/rs976552782?annotation-mode=germline
Human Phenotypes: Gait imbalance, Dysmetric saccades, Postural instability, Gaze-evoked nystagmus, Abnormality of ocular smooth pursuit, Action tremor, Hyperacusis, Progressive cerebellar ataxia, Insomnia, Poor coordination, Visual hallucinations, Impaired smooth pursuit, Dysdiadochokinesis, Abnormality of saccadic eye movements, Depression, Oculomotor apraxia
https://maayanlab.cloud/archs4/gene/IFT172


rs746816423 7-151072063-G-A my mother has it too
Missense
SLC4A2
23 out of 1,613,906 (0.0014125%)
DANN: 0.9995332606326633
CADD: 28.2



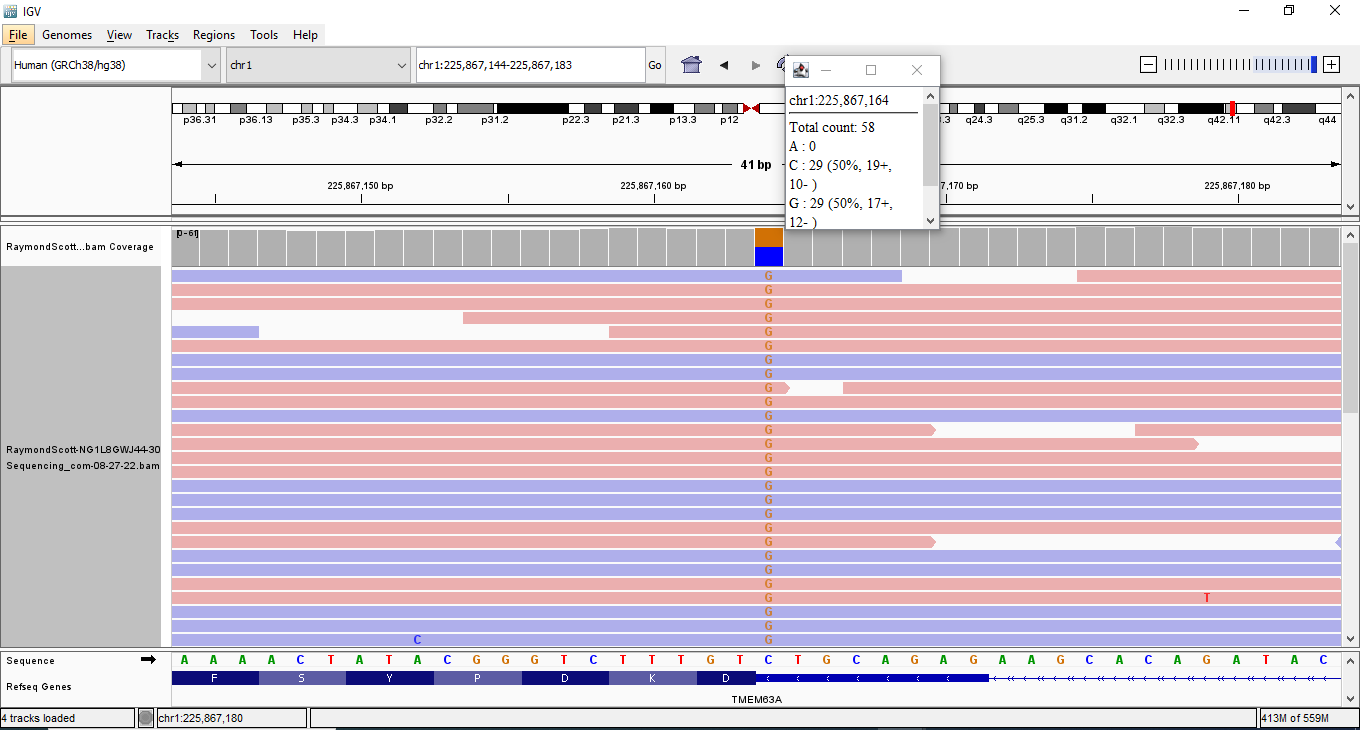
rs966645435 C/G my mother has it too
Splice Acceptor
TMEM63A
CADD: 32.0
Metascore 3 Pathogenic moderate
https://reg.clinicalgenome.org/redmine/projects/registry/genboree_registry/by_caid?caid=CA38540925
https://gnomad.broadinstitute.org/variant/1-225867164-C-G?dataset=gnomad_r4
https://varsome.com/variant/hg38/rs966645435?annotation-mode=germline
Human Phenotypes: Ataxia, Dysmetria, Specific learning disability, Intention tremor, Delayed gross motor development, Abnormality of movement, Tremor, Abnormality of coordination, Tremor, Motor delay, Abnormality of the nervous system, Abnormal central motor function, Action tremor, Abnormality of eye movement, Neurodevelopmental abnormality, Neurodevelopmental delay, Abnormal nervous system physiology, Kinetic tremor, Nystagamus
https://www.genecards.org/cgi-bin/carddisp.pl?gene=TMEM63A
Intention tremor, Abnormal auditory evoked potentials, Slow saccadic eye movements, Spastic gait, Dysmetria, Clumsiness
https://maayanlab.cloud/archs4/gene/TMEM63A
.png)


.png)

text mining disease associations include Apraxia, Episodic Ataxia
https://www.genecards.org/cgi-bin/carddisp.pl?gene=CEP290
Human Phenotypes: Photophobia, Gait imbalance, Broad-based gait, Poor coordination, Specific learning disability, Progressive cerebellar ataxia, Gaze-evoked nystagmus, Oculomotor apraxia
https://maayanlab.cloud/archs4/gene/CEP290
https://gene.sfari.org/database/human-gene/CEP290
.png)


.png)

.png)

.png)


Comments