Rare Nonsense Mediated Decay-Escaping Stop Gained Variants
This is a blog post about rare Nonsense Mediated Decay-escaping stop gained variants.
A stop-gained variant, also known as a nonsense mutation, is a type of genetic change where a single nucleotide substitution introduces a premature termination codon (PTC) within the coding sequence of a gene. This leads to early termination of translation, producing a truncated protein. Depending on the location of the PTC and the structure of the affected transcript, the variant may trigger nonsense-mediated decay (NMD) or escape it, making the biological impact isoform-specific and context-dependent.
Nonsense-mediated decay–escaping (NMD-escaping) describes a situation where a transcript containing a premature termination codon (PTC) avoids degradation by the cell’s NMD pathway and remains stable enough to be translated. While NMD typically prevents the production of truncated proteins by degrading transcripts with early stop codons, escape occurs when the PTC is located in the last exon or within ~50–55 nucleotides upstream of the final exon-exon junction. Additional transcript-specific features—such as exon structure, 3′ UTR length, and splicing—can also influence escape. The resulting protein may be benign, harmful, or functionally neutral, depending on domain context and biological role, making isoform-aware interpretation essential.
Pathogenic Rett and Angelman-like Disorders Variant Curation Expert Panel
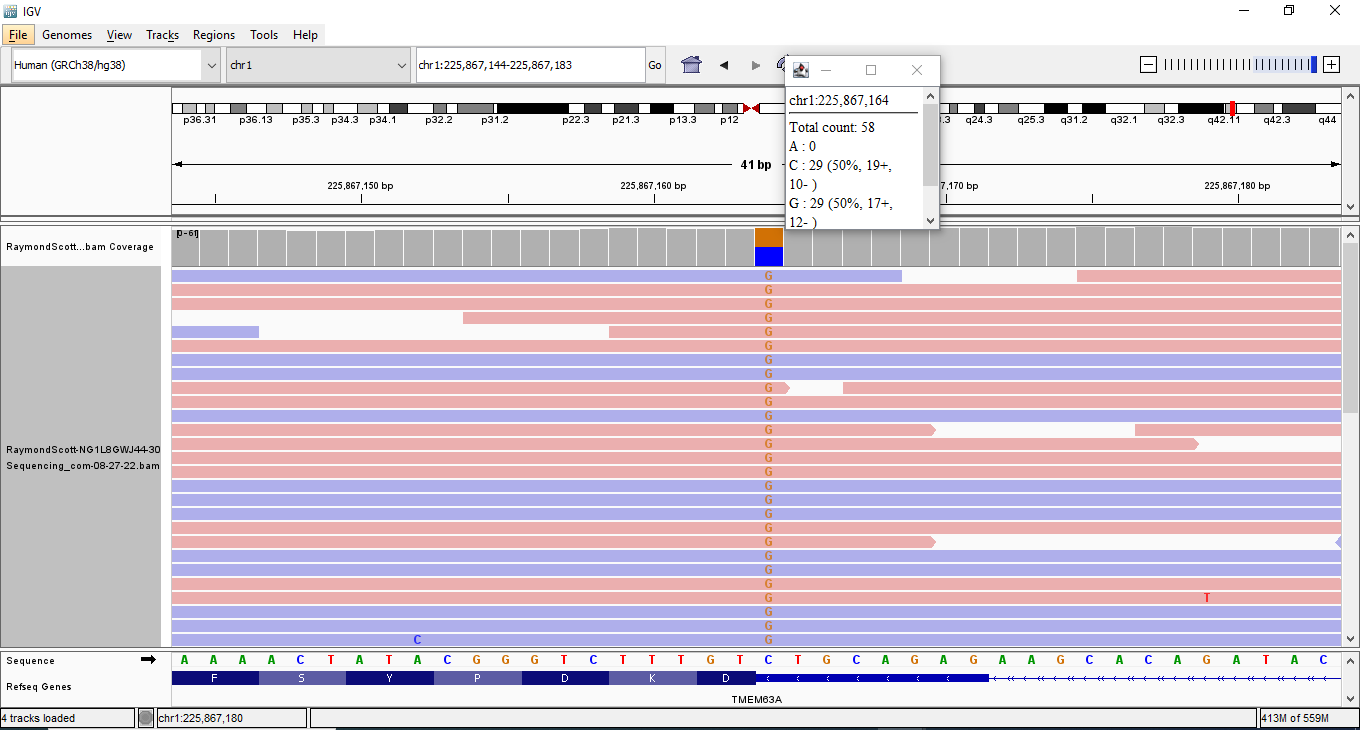
NM_001323289.2(CDKL5):c.2842C>T (p.Arg948Ter)
rs1555955296
chrX:g.18628716C>T
The following is from submission in ClinVar
Comment:
The p.Arg948X variant in CDKL5 has been reported in the hemizygous state in a young male with infantile spasms, atonic seizures, developmental delays, and regression (Schoch et al, 2020) and in the heterozygous state in a young female with infantile spasms, epilepsy, and developmental delay (Broad Institute Rare Genomes Project). In both individuals the variant was found to be de novo through trio whole genome sequence analysis. This variant has also been reported by a clinical laboratory in ClinVar (Variation ID 482247) and is absent from large population studies. This variant is present in the NM_001323289.2 transcript, which is confirmed to be expressed in the brain in both humans and mice (Hector RD et al 2016). This nonsense variant leads to a premature termination codon at position 948. This alteration occurs within the last exon and is, therefore, likely to escape nonsense mediated decay (NMD) and result in a truncated protein. However, identification of other de novo truncating variants in this region suggest the region is critical for protein function. In summary, this variant meets criteria to be classified as pathogenic for X-linked dominant early infantile epileptic encephalopathy based on case observations, de novo occurrence, absence from large population studies, and predicted impact on protein. ACMG/AMP Criteria applied: PS2, PVS1_Strong, PM2_Supporting. (less)
Early infantile epileptic encephalopathy with suppression bursts
Affected status: unknown
Allele origin: germline
Laboratory for Molecular Medicine, Mass General Brigham Personalized Medicine
Accession: SCV004848397.1
First in ClinVar: Apr 20, 2024
Last updated: Apr 20, 2024
I checked the in-silico predictions at Varsome website. The overall in-silico predictions show it as being benign.
BayesDel addAF - Benign Strong
BayesDelnoAF - Benign Moderate.
CADD - 16.34 - low for a Pathogenic variant (especially for a Stop Gained variant). I use a minumum of 20.
DANN - 0.80557297479159784 suggesting benign

I checked the variant using Variant Effect Predictor (VEP). It affects two transcripts with the following properties:
• Transcript 1: 18/18 exons
• cDNA position: 3092
• CDS position: 2842
• Protein position: 948
• Transcript 2: 19/19 exons
• cDNA position: 3215
• CDS position: 2965
• Protein position: 989
• Consequence: Stop-gained in the final coding exon of both transcripts
• NMD status: Escapes nonsense-mediated decay
This CDKL5 variant affects two structurally shortened transcripts, one including 18 of the gene’s 24 exons, and the other including 19. Both transcripts are incomplete even before the variant occurs. The stop-gained variant then truncates the remaining tail, compounding the structural loss. As a result, the expressed protein is malformed not only by premature termination, but by originating from an already incomplete blueprint.
The fact that this is a shortened, NMD-escaping transcript amplifies the biological impact. It expresses a malformed protein that wouldn’t exist if the variant triggered NMD—making it a potential driver of pathogenicity.
PVS1_Strong is justified not by generic LoF, but by domain-level disruption in an expressed isoform.
• PS2 (de novo) adds weight, but the biological plausibility hinges on transcript-aware reasoning is now confirmed.
• In-silico benignity is overridden by functional domain loss, which most predictors don’t model.

NM_014840.3(NUAK1):c.1369A>T (p.Lys457Ter)
chr12:g.106067419T>A
Stop Gained NMD-escaping variant
not found in any population
BayesDel addAF - Pathogenic Strong
BayesDelnoAF - Pathogenic Strong
CADD - 38 indicating highly deleterious
DANN - 0.9961 indicating Damaging
A NMD-escaping stop-gained variant in NUAK1 introduces a premature stop codon in the final coding exon of a transcript, allowing production of a truncated protein. NUAK1 encodes a serine/threonine kinase involved in cell survival and metabolic stress response. Truncation may impair signaling pathways relevant to hypoxia adaptation, tumor progression, or neural resilience.

I checked the variant using Variant Effect Predictor (VEP). It affects one transcript with the following properties:
• Transcript structure: 7/7 exons
• cDNA position: 1670
• CDS position: 1369
• Protein position: 457
• Consequence: Stop-gained in the final coding exon
• NMD status: Escapes nonsense-mediated decay
This NUAK1 variant affects a structurally shortened transcript that includes only 7 of the gene’s 12 exons. The transcript is incomplete even before the variant occurs. The stop-gained variant then truncates the remaining tail, compounding the structural loss. As a result, the expressed protein is malformed not only by premature termination, but by originating from an already incomplete blueprint.


NM_014853.3(SGSM2):c.3010C>T (p.Arg1004Ter)
chr17:g.2379146C>T
rs867998099
Stop Gained NMD-Escaping variant
3 out of 1,614,054 (0.0001859%) GnomAD
2 out of 490,784 (0.0004%) AllofUs
2 out of 264,690 (0.0008%) TOPMED
2 out of 7234 (0.03%) Korea4K
1 out of 1832 (0.05%) Korea1K
1 out of 16,760 (00.006%) 8.3KJPN
BayesDelnoAF - Pathogenic Strong
CADD - 40 indicated highly deleterious
DANN - 0.9978 suggesting Damaging

• Transcript structure: 2/2 exons
• cDNA position: 152
• CDS position: 154
• Protein position: 52
• Consequence: Stop-gained in the final coding exon
• NMD status: Escapes nonsense-mediated decay
This SGSM2 variant affects a structurally shortened transcript that includes only 2 of the gene’s 24 exons. The transcript is incomplete even before the variant occurs. The stop-gained variant then truncates the remaining tail, compounding the structural loss. As a result, the expressed protein is malformed not only by premature termination, but by originating from an already incomplete blueprint.



Comments